Computational Virology
Treatment of HIV infection is based on combinations of antiviral drugs. To identify the optimal drug combination for each patient, we develop computational methods based on the genetic composition of the virus population of the patient. We predict the probability that the virus will develop escape mutations that would result in drug resistance and eventually in treatment failure. This personalized medicine approach allows for defining drug combinations that are optimized for each individual patient.
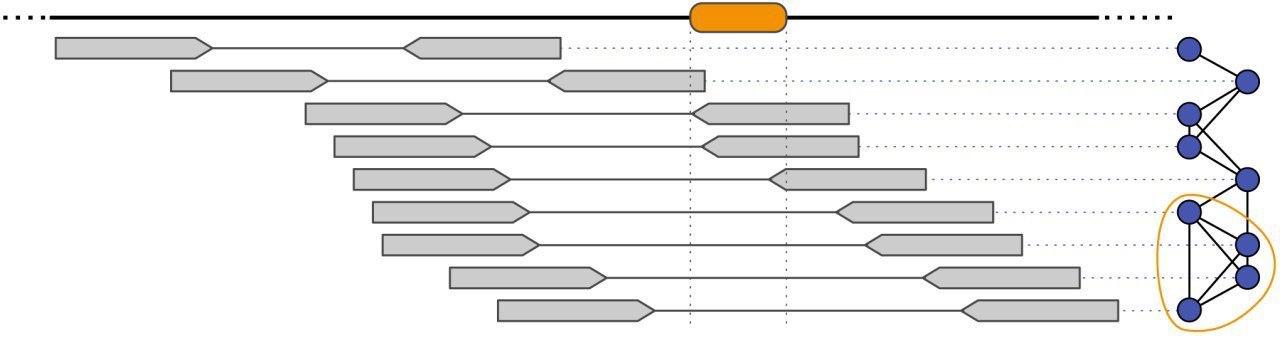
In order to estimate the genetic composition of an intra-host virus population, we have developed several computational approaches for the analysis of ultra-deep sequencing data. This challenging task, known as the viral quasispecies assembly problem, involves the assembly of all sequencing reads into an unknown number of unknown viral haplotype sequences.
In order to make ultra-deep sequencing of virus populations accessible to a broad range of biomedical users, we develop and actively maintain external page V-pipe, a bioinformatics pipeline for the analysis of viral high-throughput sequencing data. V-pipe supports the reproducible analysis of genomic diversity in intra-host virus populations, which is involved in viral pathogenesis and virulence, and enables diagnostic applications.
Selected references:
- external page Viral Quasispecies Assembly via Maximal Clique Enumeration
Töpfer et al., PLoS Comput Biol, 2014
- external page Single-Cell RNA-Seq Reveals Transcriptional Heterogeneity in Latent and Reactivated HIV-Infected Cells
Golumbeanu et al., Cell Rep, 2018 - Host factor prioritization for pan-viral genetic perturbation screens using random intercept models and network propagation
Dirmeier et al., PLoS Comput Biol, 2020