Research
Research in the Computational Biology Group aims to advance the rational design of medical interventions in complex and evolving biological systems, such as cancerous tumors and virus populations. We employ an array of methods and approaches: developing models and algorithms for the statistical analysis of high-throughput molecular data; reconstructing and analyzing biological networks; predicting the effect of perturbations; and designing evolutionary models of rapidly adapting cells and pathogens. Our methodologies are applied in several AI-driven personalized medicine initiatives, with a particular emphasis on oncology and virology.
Computational Oncology
The goal of our cancer-related research is to support the diagnosis and treatment of cancer. Our research topics include the analysis, integration, and interpretation of molecular profiles of cancer cell populations, and across patient cohorts. For example, we have developed methods for mutation calling, for reconstructing the evolutionary history of a tumor, for discriminating driver from passenger mutations, and for identifying mutually exclusive cancer mutations which are likely to be critical for tumor growth and hence candidates for medical interventions. We are part of the Tumor Profiler Center, where we develop methods for single-cell genomics and transcriptomics data from cancerous tumor cells and for data integration across various single-cell technologies. These methods can improve patient outcomes by enabling early detection and diagnosis and by facilitating the development of personalized treatment plans tailored to individual genetic and clinical profiles.
- external page Kevin Rupp, Andreas Lösch, Y. Linda Hu, Chenxi Nie, Rudolf Schill, Maren Klever, Simon Pfahler, Lars Grasedyck, Tilo Wettig, Niko Beerenwinkel, and Rainer Spang. Modeling metastatic progression from cross-sectional cancer genomics data. Bioinformatics, 40(suppl 1):i140–i150, 2024. (ISMB 2024)
- external page Xiang Ge Luo, Jack Kuipers, Kevin Rupp, Koichi Takahashi, and Niko Beerenwinkel. Bayesian inference of fitness landscapes via tree-structured branching processes. Bioinformatics. 2025.
- external page Nicola Miglino, Nora C. Toussaint, Alexander Ring et al. Feasibility of multiomics tumor profiling for guiding treatment of melanoma. Nature Medicine 31, 2430–2441 (2025).
- external page Kiyomi Morita, Feng Wang, Katharina Jahn, et al. Clonal evolution of acute myeloid leukemia revealed by high-throughput single-cell genomics. Nat Commun 11, 5327 (2020).
- external page Fritz Bayer, Marco Roncador, Giusi Moffa et al. Network-based clustering unveils interconnected landscapes of genomic and clinical features across myeloid malignancies. Nature Communications 16, 4043 (2025).
- external page Jack Kuipers, Katharina Jahn, Benjamin J. Raphael, and Niko Beerenwinkel. Single-cell sequencing data reveal widespread recurrence and loss of mutational hits in the life histories of tumors. Genome Research, 27:1885-1894, 2017.
- external page David Gremmelspacher, Johannes Gawron, Barbara M. Szczerba et al. Phylogenetic inference reveals clonal heterogeneity in circulating tumor cell clusters. Nat Genet 57, 1357–1361 (2025).
Computational Virology
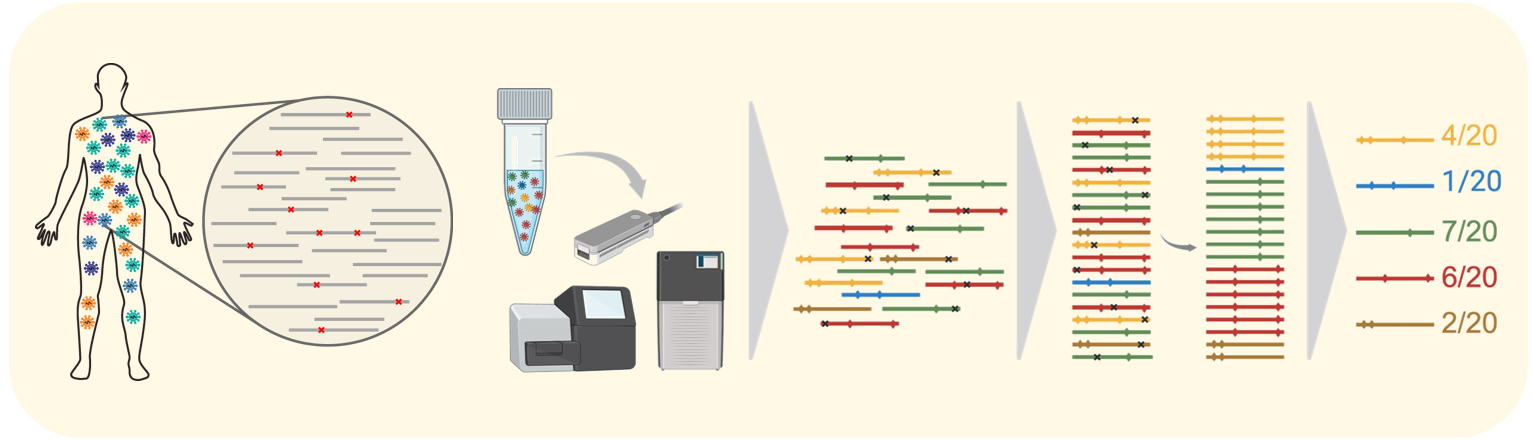
Our research in computational virology aims at identifying optimal personalized antiviral drug combinations tailored to the genetic composition of each patient´s virus population. We predict the probability that the virus will develop genetic escape mutations, which could lead to drug resistance and eventually result in treatment failure. In order to estimate the genetic makeup of a virus population within a host, we have developed computational approaches for the assembly of sequencing reads into viral haplotype sequences. The other major focus of our computational virology research involves analyzing wastewater samples to detect the presence of pathogens, such as SARS-CoV-2, RSV, and Influenza, to obtain population-level epidemiological information. Our computational virology tools are integrated into V-pipe, our bioinformatics pipeline designed for the reproducible analysis of mixed virus samples.
- external page Lara Fuhrmann, Kim Philipp Jablonski, Ivan Topolsky, Aashil A. Batavia, Nico Borgsmüller, Pelin Icer Baykal, Matteo Carrara, Chaoran Chen, Arthur Dondi, Monica Drăgan, David Dreifuss, Anika John, Benjamin Langer, Michal Okoniewski, Louis du Plessis, Uwe Schmitt, Franziska Singer, Tanja Stadler, and Niko Beerenwinkel. V-pipe 3.0: a sustainable pipeline for within-sample viral genetic diversity estimation. GigaScience, 13:giae065, 2024.
- external page Lara Fuhrmann, Benjamin Langer, Ivan Topolsky, and Niko Beerenwinkel. VILOCA: Sequencing quality-aware haplotype reconstruction and mutation calling for short- and long-read data. NAR Genomics and Bioinformatics, 2024.
- external page Katharina Jahn, David Dreifuss, Ivan Topolsky, Anina Kull, Pravin Ganesanandamoorthy, Xavier Fernandez-Cassi, Carola Bänziger, Elyse Stachler, Lara Fuhrmann, Kim Philipp Jablonski, Chaoran Chen, Catharine Aquino, Tanja Stadler, Christoph Ort, Tamar Kohn, Timothy R. Julian, and Niko Beerenwinkel. Early detection and surveillance of SARS-CoV-2 genomic variants in wastewater using COJAC. Nature Microbiology, 7:1151–1160, 2022.
- external page Armin Töpfer, Tobias Marschall, Rowena A. Bull, Fabio Luciani, Alexander Schönhuth, and Niko Beerenwinkel. Viral quasispecies assembly via maximal clique enumeration. PLoS Computational Biology, 10(3):e1003515, 2014. (RECOMB 2014)
- external page David Dreifuss, Jana S. Huisman, Johannes C. Rusch et al. Estimated transmission dynamics of SARS-CoV-2 variants from wastewater are unbiased and robust to differential shedding. Nature Communications 16, 7456 (2025).
- external page Osvaldo Zagordi, Rolf Klein, Martin Däumer, and Niko Beerenwinkel. Error correction of next-generation sequencing data and reliable estimation of HIV quasispecies. Nucleic Acids Research, 38(21):7400–7409, 2010.