Genomic Epidemiology
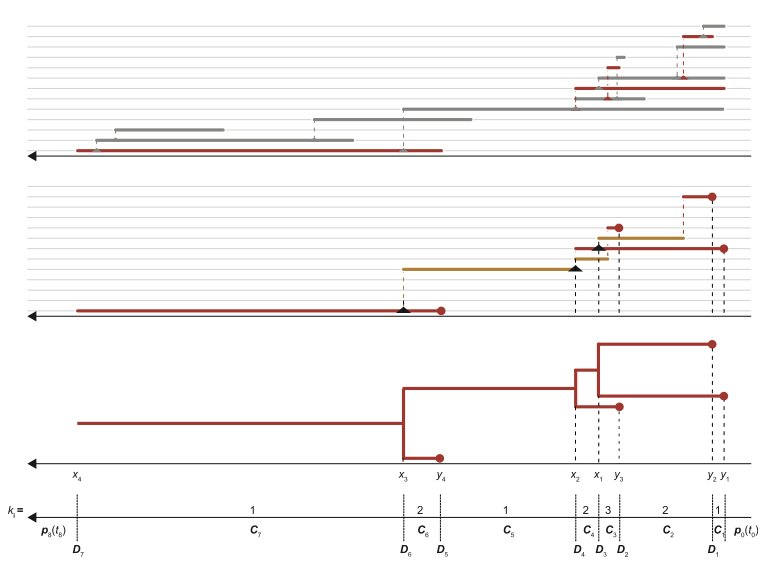
Many pathogens show a substantial amount of genetic variation over the course of an epidemic. This is especially true for pathogens such as RNA viruses, which have short generation times and high mutation rates. In this case, epidemiological and evolutionary dynamics occur on the same timescale and we can use phylogenetic methods to reconstruct a phylogeny from sampled pathogen sequences. This phylogeny represents an approximation to the true transmission tree of the epidemic, as illustrated above.
By incorporating classic epidemiological models into phylogenetic inference we are able to quantify the epidemiological dynamics (i.e. transmission and recovery) of the pathogen. Such an approach allows us to infer factors responsible for the epidemic spread, such as the importance of population structure or superspreaders. We can also infer the effectiveness of public health policies in bringing the epidemic under control by looking at changes in the epidemiological parameters over time. These inferences are important for a well-informed public health response. We currently focus on obtaining a better understanding of the spread of SARS-CoV-2, HIV, HCV, Influenza, Ebola, and Tuberculosis.