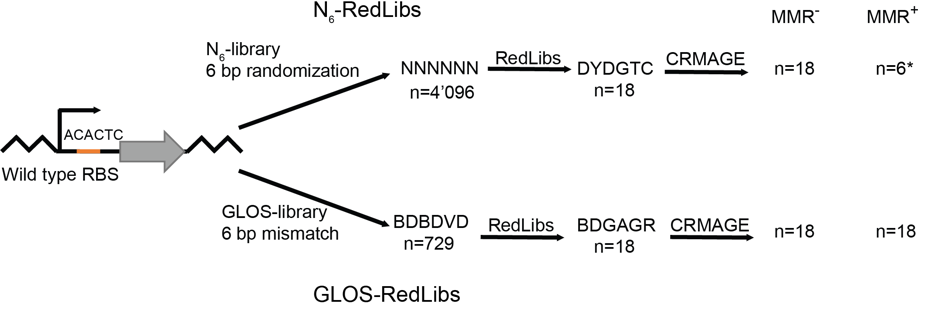
Genome engineering and genome editing
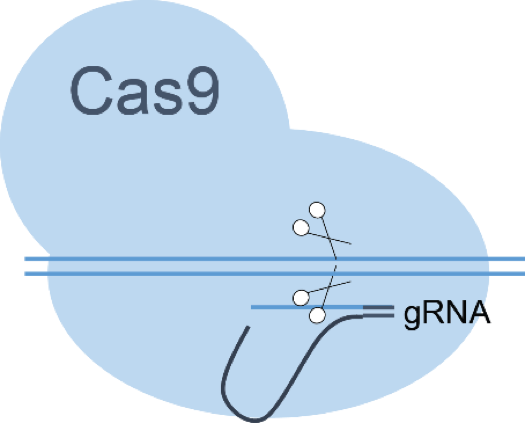
High-performing producer strains are essential for the biotechnological production of fine, specialty, and commodity chemicals. A suitable prokaryotic producer organism must not only be capable of overproduction of the compound of interest, but it also needs a stable genotype. This is usually tantamount to placing genetic information on the bacterial chromosome and most often requires methods that allow rapidly modification of multiple genomic functions. We are mainly using MAGE-based genome editing with CRISPR/Cas9 counter-selection. Applied examples are RBS optimization and translational or full gene knockouts for metabolic engineering, internal genomic tagging for protein knockdowns, and genomic integration of fluorescent proteins for biomass markers.